



Specifications:
Further Information
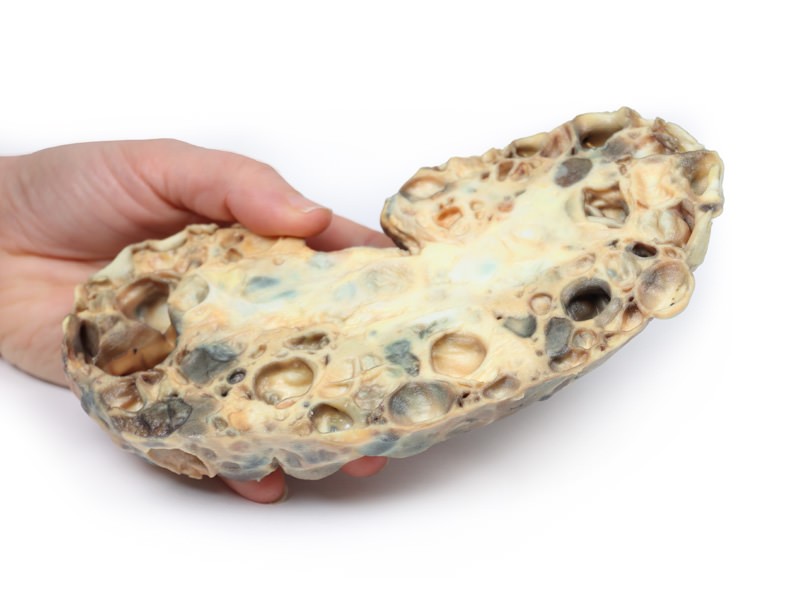
Adult polycystic kidney disease (APKD) is an autosomal dominant disorder characterised by the presence of multiple cysts within the renal parenchyma. The cysts develop from altered renal tubule epithelium. The cysts expand destroying the glomeruli, causing ischaemia, pressure atrophy, and eventually leading to renal failure. APKD occurs in 1 in 40 to 1000 live births. Mutations in the PKD1 gene on chromosome 16p13.3 and PKD2 gene on chromosome 4q21 have been described as causal mutations. These code for membrane proteins polycystin 1 and 2, respectively. Patients with PKD1 mutation are more common and have a more severe phenotype. End stage renal disease (ESRD) occurs at a mean age of 74.0 in PKD2 versus 54.3 years in PKD1. Common symptoms of APKD include haematuria from haemorrhage into cysts and pain or a sensation of dragging from the expansion of cysts and kidney enlargement. Many patients remain asymptomatic until features of renal failure occur such as proteinuria, polyuria, hypertension and uraemia. Extrarenal manifestations of the disease include intracranial ‘berry’ aneurysms, hepatic and pancreatic cysts, as well as mitral valve prolapse and other types of cardiac valve disease. Renal ultrasound is the most common investigation used to diagnose APKD. CT- and MRI scans may also be used as diagnostic tools. Patients with a positive family history of APKD can be offered screening renal US scans and genetic testing in some cases. Treatment involves renal replacement therapy for ESRD and renal transplant (if a donor can be found). Ultimately, over one-third of patients die from renal failure and one-third from coronary or hypertensive heart disease. Approximately 1% of patients die from subarachnoid haemorrhage, because of berry aneurysm rupture (as in this case). Remaining deaths are due to unrelated causes.
Catalogue: PDF
See more products OpenMedis brand.



Reviews
There are no reviews yet.